
近日,国家南亚标准化(成都)研究中心研究人员从监管机构、法律法规、清关流程与材料和相关标准等四个方面,对印度、巴基斯坦、孟加拉国、尼泊尔、不丹、斯里兰卡等六国的防护用品市场准入要求进行了探究。现将阶段性研究成果公布如下,以期对各相关方有所助益。
印度
监管机构
中央药品标准控制机构(Central Drugs Standard Control Organization,CDSCO),是印度卫生与福利部(Ministry of Health and Family Welfare)下属的监管医疗器械进口、生产和销售的重要部门。中央药品标准控制机构曾经是管理药品、诊断试剂和化妆品的标准、法规、生产和进口的部门。现在,CDSCO的职能是制定药品、疫苗、血液制品和注射液的标准、法规。同时该机构增加了监管医疗器械的职能,为了加强对医疗器械的监督管理。
印度认证机构国家认可委员会(NABCB)由印度工商部下的印度质量委员会建立的,其目的是作为国家认可机构认证公告机构。国家认可委员会确定公告机构的合格评定活动及认可标准,制订公告机构的认可准则及程序,并定期对公告机构进行审核,以评估其是否符合医疗器械规则。
法律法规
在“印度制造”计划的支持下,2017年1月31日,卫生和家庭福利部( Ministry of Health & Family Welfare)颁布了《医疗器械条例,2017》,自2018年1月1日起生效,并于2018年6月1日颁布了《医疗器械条例补充条款,2018》。在2017年《医疗器械条例》实施之前,印度根据《药品和化妆品法,1945》将医疗器械作为药物(制药产品)进行监管,因此,该条例规定医疗器械应与药物分开。
新规符合全球协调工作组(GHTF)框架,符合最佳国际惯例,并根据风险对医疗器械进行分类。根据新规,医疗器械被分类为A级(低风险)、B级(低至中度风险)、C级(中至高风险)和D级(高风险)。2017年11月1日,药品管理局(Drugs Controller General,DCGI)根据《医疗器械条例,2017》的规定,通报了351种医疗器械和247种体外诊断医疗器械及其风险等级。这份清单详细列出了之前根据《药品和化妆品法》通报的15类医疗器械和8种物质。
新规的制定是为了促进印度国内制造业的发展,并规范该地区的进口和制造业。此外,新的医疗器械规则中也引入了公告机构的检查。《医疗器械条例,2017》介绍了由公告机构进行的第三方合格评定。印度认证机构国家认可委员会(NABCB)已被被中央许可证颁发机构(CLA)确定为授权认可机构。公告机构将对A类和B类医疗器械制造商的质量管理体系进行核查和评估,如有需要,还将被要求协助对C类和D类医疗器械进行监管。制造商的质量管理体系必须符合ISO 13485。
清关流程与材料
目前,印度按照风险级别将医疗器械产品分为A、B、C、D四类。A类产品不需要许可证,但其自愿申请须由州许可当局(SLA)许可;B类设备需要(SLA)审批;C类和D类设备需要中央许可局(CLA)审批。医疗器械产品分为管制类型与非管制类型。
1、非管制类型:
目前,印度只有40-50个医疗器械需要注册。对于所有其他不需要注册的医疗器械,制造商应从DCGI获得无异议证书(NOC)。NOC是来自DCGI的一封信,声明该产品不需要注册,可以自由进口到印度。
2、管制类型:
医疗器械要在印度上市,需要申请医疗器械注册证,注册证书有效期为三年,需要在证书有效期九个月前提交重新注册申请。
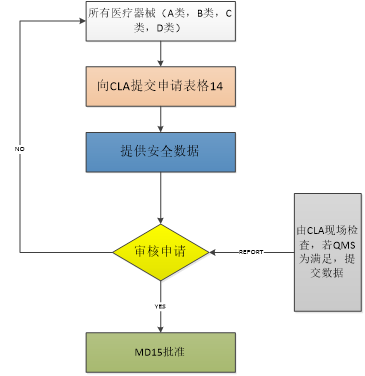
图1:医疗器械进口许可证审批
所有医疗器械产品进口都需要获得印度药品管制局ADC 的无异议证书(No Objection Certificate,NOC)。协助药品管制员(ADC)会询问药品的进口许可证。如进口原材料,那么协助药品管制员(ADC)会要医疗器械产品的生产许可证。其他进口清关重要还有商业发票、装箱单、货运证书、保险证书、原产地证书以及空运单/提货单。
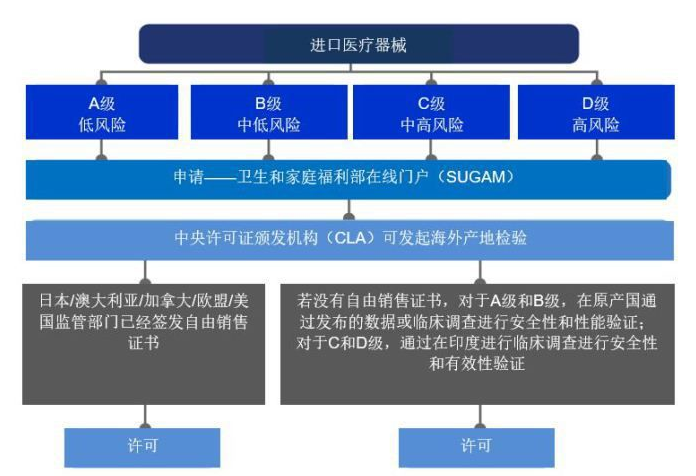
图2:医疗器械进口流程
相关标准
印度医疗器械产品遵循的标准总结如下:
-制造商必须遵循中央政府为医疗器械专门制定的或BIS制定的标准;
-若未制定任何医疗器械的相关标准,则该器械应符合国际标准化组织(ISO)或国际电工委员会(IEC)或任何其他药典标准制定的标准;
-如果没有上述标准类别,则应采用经验证的制造商标准即客户标准。
印度标准局BIS针对包括基础和生产工程、化工、城建、电子和通信、食品与农业、机械工程、管理和体系、医药设备和医院规划等15个领域设立了相应的16个分部理事会,将标准制定工作分配给涉及不同的技术范围的分部理事会。其中,口罩、手套以及防护服等相关标准主要涉及医药设备和医院规划(Medical Equipment and Hospital Planning Department,MHD)分部委员会,化工(Chemical Department ,CHD)以及纺织(Textile Department ,TXD)分部委员会也制定了少量标准。
表1: 印度防护产品标准
类别 | 标准号 | 标准名称 |
口 罩 | IS 16288 : 2014 (Reaffirmed Year : 2019 ) | 医用纺织品 外科口罩细菌过滤效率评估方法 |
IS 16289 : 2014 (Reaffirmed Year : 2019 ) | 医用纺织品 外科口罩规范 | |
IS 14166 : 1994 (Reaffirmed Year : 2014 ) | 呼吸防护装置:全面罩 | |
IS 14746 : 1999 (Reaffirmed Year : 2014 ) | 呼吸防护装置-半面罩和四分之一面罩 | |
IS 9473 : 2002 (Reaffirmed Year : 2014 ) | 呼吸防护装置-防止颗粒的过滤半面罩 | |
IS 9623 : 2008 (Reaffirmed Year : 2018 ) | 呼吸防护装置的选择、使用和维护-规程 | |
IS 6190 : 1971 (Reaffirmed Year : 2014 ) | 麻醉面罩规范 | |
防护服 | IS 16548-2016 | 预防传染性病原体的衣服-耐干微生物渗透性的试验方法 |
IS 16545-2016 | 防止接触血液和体液的衣服-血传播致病菌对防护服材料渗透性的测定-Phi-X174噬菌体试验方法 | |
IS 16546-2016 | 防止接触血液和体液的衣服-血液和体液渗透性测定防护服材料的渗透性-合成血液的试验方法 | |
IS 15758 : Part 1 : 2007 (Reaffirmed Year : 2018 ) | 纺织品-防护服:第1部分测定火焰中热传递的方法 | |
IS 15758 : Part 2 : 2007 (Reaffirmed Year : 2018 ) | 纺织品-防护服:第2部分暴露于辐射热源时材料组件的评定 | |
IS 15758 : Part 3 : 2007 (Reaffirmed Year : 2018 ) | 纺织品-防护服:第3部分材料耐液体渗透的试验方法 | |
IS 15758 : Part 4 : 2007 (Reaffirmed Year : 2018 ) | 纺织品-防护服:第4部分有限火焰蔓延的试验方法 | |
IS 15758 : Part 5 : 2007 (Reaffirmed Year : 2018 ) | 纺织品-防护服:第5部分材料对熔融金属飞溅的阻力的评估 | |
IS 15071 : 2002 (Reaffirmed Year : 2014 ) | 化学防护服-规范 | |
护目镜 | IS 5983 : 1980 (Reaffirmed Year : 2018 ) | 眼睛保护器 |
手 套 | IS 4148 : 1988 (Reaffirmed Year : 2016 ) | 医用橡胶手套规范 |
IS 7180 : 1973 (Reaffirmed Year : 2019 ) | 一次性人工授精手套 | |
IS 15354-1-2018 | 一次性医疗检验手套 第1部分 橡胶乳胶或胶浆制手套 第1部分 规范(Mhd 12) | |
IS 15354-2-2018 | 一次性医疗检验手套 第2部分 聚氯乙烯手套规范(Mhd 12) | |
IS 13422 : 1992 (Reaffirmed Year : 2013 ) | 一次性医用橡胶手套规范 | |
消毒剂 | IS 1061-2017 | 酚醛型消毒液体 规范(第五版) |
IS 4094 : 1967 (Reaffirmed Year : 2016 ) | 中文名:手术钳、消毒箱、奇特尔模式 | |
IS 10758 : 1983 (Reaffirmed Year : 2019 ) | 除臭-消毒液 | |
IS/ISO 15883 :Part 1: 2006 | 洗涤消毒器 第1部分 一般要求、术语、定义和测试 | |
IS/ISO 15883 :Part 2 : 2006 | 洗涤消毒剂 第2部分 外科器械、麻醉设备、餐具、听筒、玻璃器皿等用热消毒洗涤消毒剂的要求和试验 | |
IS/ISO 15883 :Part 3 : 2006 | 洗涤-消毒器:第3部分使用对人体废物容器进行热消毒的洗衣机消毒器的要求和试验 | |
IS/ISO 11607-1-2006 | 终端消毒医疗设备的包装 第1部分 材料、无菌屏障系统和包装系统的要求 | |
IS/ISO 11607-2-2006 | 终端消毒医疗设备包装 第2部分 包装、密封和装配过程检验要求 | |
IS 10150 : 1981 (Reaffirmed Year : 2016 ) | 医疗产品灭菌指南 | |
IS/ISO 10993:Part7:2008 | 医疗器械的生物评价:第7部分环氧乙烷灭菌残留物 | |
IS/ISO 11140: Part4:2007 | 卫生保健产品的灭菌-化学指标:第4部分第2类指标作为蒸汽渗透检测的鲍威和狄克式试验的替代方法。 | |
IS/ISO 11140: Part5:2007 | 卫生保健产品的灭菌-化学指标:第5部分第2类指标鲍威和迪克-空气去除试验类型 | |
IS/ISO 11137: Part1:2006 | 卫生保健产品的灭菌-辐射:第1部分医疗器械灭菌过程的开发、验证和常规控制的要求 | |
IS/ISO 11135: 2014 | 卫生保健产品的灭菌-环氧乙烷-医疗器械灭菌过程的开发、验证和常规控制的要求 | |
IS/ISO 11137:Part2:2013 | 卫生保健产品的灭菌-辐射:第2部分:灭菌剂量的确定 | |
IS/ISO 11140:Part3:2007 | 保健产品的灭菌-化学指标:第3部分鲍威和迪克蒸汽渗透试验用第2类指标体系 | |
IS/ISO 14161:2009 | 卫生保健产品的灭菌-生物指标-结果选择、使用和解释指南 | |
IS/ISO 11137:Part3:2017 | 卫生保健产品的灭菌-辐射第3部分开发、验证和常规控制的剂量学方面的指南 | |
IS/ISO 11138:Part1:2017 | 卫生保健产品的灭菌-生物指标第1部分通用要求 | |
IS/ISO 11138:Part2:2017 | 卫生保健产品的灭菌-生物指标第2部分环氧乙烷灭菌过程的生物指标 | |
IS/ISO 11138:Part3:2017 | 卫生保健产品的灭菌-生物指标第3部分湿热灭菌过程的生物指标 | |
IS/ISO 11138:Part4:2017 | 卫生保健产品的灭菌-生物指标第4部分干热灭菌过程的生物指标 | |
IS/ISO 11138:Part5:2017 | 卫生保健品的灭菌-生物指标第5部分低温蒸汽和甲醛灭菌过程的生物指标 | |
测温计 | IS 15113 : 2002 (Reaffirmed Year : 2017 ) | 带最大装置的临床电子体温计-规格 |
IS 3055 : Part 1 : 1994 (Reaffirmed Year : 2016 ) | 临床体温计:第1部分实心茎型-规范 | |
IS 3055 : Part 2 : 2004 (Reaffirmed Year : 2014 ) | 临床体温计:第2部分封闭式秤-规范 | |
IS 4529 : 1968 (Reaffirmed Year : 2018 ) | 医用温度计用玻璃管规范 | |
IS 4610 : 1968 (Reaffirmed Year : 2016 ) | 通用和标准温度计用玻璃管规范 | |
IS 80601 : Part 2 : Sec 56 : 2017 | 医用电气设备:第2部分基本安全和基本性能的特殊要求:第56节体温测量用临床温度计 | |
呼吸机(器) | IS/ISO 5356 : Part 1 : 2015 | 麻醉和呼吸设备:锥形连接器:第1部分锥形和插座(第一次修订) |
IS/ISO 5356 : Part 2 : 2012 | 麻醉和呼吸设备:锥形连接器:第2部分螺旋螺纹承重连接器 | |
IS/ISO 5361 : 1999 (Reaffirmed Year : 2017 ) | 麻醉和呼吸设备:气管导管和连接器 | |
IS/ISO 5364 : 2008 (Reaffirmed Year : 2014 ) | 麻醉和呼吸设备口咽呼吸道 | |
IS/ISO 5366 : PART 1 : 2000 (Reaffirmed Year : 2017 ) | 麻醉和呼吸设备:气管造口管:第1部分成人用管和连接器 | |
IS 7409 : Part 2 : 1994 (Reaffirmed Year : 2016 ) | 麻醉和呼吸设备:锥形连接器:第2部分:螺纹承重连接器 | |
IS 8347 : 2008 (Reaffirmed Year : 2018 ) | 呼吸防护用品:部件的定义、分类和命名 | |
IS 10245 : Part 2 : 1994 (Reaffirmed Year : 2017 ) | 呼吸保护装置:呼吸器:第2部分开路呼吸装置 | |
IS/ISO 11712 : 2009 (Reaffirmed Year : 2014 ) | 麻醉和呼吸设备 | |
IS 13200 : 2015 | 麻醉和呼吸设备:词汇(第一版) | |
IS 14138 : Part 1 : 1994 (Reaffirmed Year : 2014 ) | 呼吸防护设备:面罩用螺纹:第1部分标准螺纹连接 | |
IS 14138 : Part 2 : 1994 (Reaffirmed Year : 2014 ) | 呼吸保护装置:面罩用螺纹:第2部分中心螺纹连接 | |
IS 14170 : 1994 (Reaffirmed Year : 2014 ) | 呼吸保护装置:吹口组件 | |
IS/ISO 15001 : 2010 | 麻醉和呼吸设备:氧兼容性(第一版) | |
IS 15322 : 2003 (Reaffirmed Year : 2014 ) | 用于呼吸防护设备的颗粒过滤器 | |
IS 15323 : 2003 (Reaffirmed Year : 2014 ) | 用于呼吸保护设备的气体过滤器和组合过滤器 | |
IS 15803 : 2008 (Reaffirmed Year : 2018 ) | 呼吸保护装置:自给式闭路呼吸器化学氧(KO2)型自产自救装置 | |
IS 17274 : Part 1 : 2019 | 呼吸保护装置:测试方法和测试设备:第1部分测定内泄漏 | |
IS 17274 : Part 2 : 2019 | 呼吸保护装置:测试方法和测试设备:第2部分测定呼吸阻力 | |
IS 17274 : Part 3 : 2019 | 呼吸保护装置:测试方法和测试设备:第3部分粒子过滤渗透的测定 | |
IS 17274 : Part 5 : 2019 | 呼吸保护装置:测试方法和测试设备:第5部分呼吸机,代谢模拟器,RPD头部和躯干,工具和验证工具 | |
IS 17274 : Part 6 : 2019 | 呼吸保护装置:测试方法和测试设备:第6部分部件和连接的机械阻力/强度 | |
IS 17274 : Part 7 : 2019 | 呼吸保护装置:测试方法和测试设备:第7部分实用性能测试方法 | |
IS 17274 : Part 8 : 2019 | 呼吸保护装置:测试方法和测试设备:第8部分RPD辅助过滤空气流量的测量 | |
IS 17274 : Part 9 : 2019 | 呼吸保护装置:测试方法和测试设备:第9部分吸入气体中二氧化碳含量的测定 | |
IS 17274 : Part 10 : 2019 | 呼吸保护装置:测试方法和测试设备:第10部分耐火性、火焰性、辐射热、耐热性 | |
IS 17274 : Part 11 : 2019 | 呼吸保护装置:测试方法和测试设备:第11部分视野确定 | |
IS 17274 : Part 12 : 2019 | 呼吸保护装置:测试方法和测试设备:第12部分呼吸体积平均功和呼吸峰值压力的测定 | |
IS 17274 : Part 13 : 2019 | 呼吸保护装置:测试方法和测试设备:第12部RPD使用再生透气气体和特殊应用矿业逃脱RPD:合并测试气体浓度,温度,湿度,呼吸,呼吸 | |
IS/ISO 80601 : Part 2 : Sec 12 : 2011 | 医疗电气设备:第2部分第12节重症监护呼吸机基本安全和基本性能的特殊要求 | |
个人防护鞋 | IS 15298 : Part 1 : 2015 | 个人防护装备:第1部分鞋类的试验方法(第二版) |
IS 15298 : Part 2 : 2016 | 个人防护装备:第2部分安全鞋 | |
IS 15298 : Part 3 : 2018 | 个人防护装备:第3部分防护鞋(第二版) | |
IS 15298 : part 4 : 2017 | 个人防护装备:第4部分职业鞋(第二版) | |
IS 15298 : Part 5 : 2004 (Reaffirmed Year : 2016 ) | 职业用安全、防护和职业鞋:第5部分附加要求和试验方法 | |
IS 15298 : Part 6 : 2004 (Reaffirmed Year : 2016 ) | 职业用安全、防护和职业鞋:第6部分安全鞋的附加规范 | |
IS 15298 : Part 7 : 2004 (Reaffirmed Year : 2016 ) | 职业用安全、防护和职业鞋:第7部分防护鞋的附加规范 | |
IS 15298 : Part 8 : 2004 (Reaffirmed Year : 2016 ) | 职业用安全、防护和职业鞋:第8部分职业鞋的附加规范 | |
质量管理体系 | IS 23485 : 2019 | 医疗器械-质量管理体系要求以及医疗器械安全性和性能的基本原则 |
表2:印度IS 16289医用纺织品外科口罩规范
性能 | 要求 | 测试方法 | |||
等级1 | 等级2 | 等级3 | IS号 | 附录 | |
细菌过滤效果%(不小于) | 95 | 98 | 98 | IS1 6288 | |
压力差Pa(不超过) | 29.4 | 29.4 | 49 | C | |
抵抗合成血液渗透(不小于mm Hg) | - | - | 120 | D | |
>100纳米乳胶球颗粒过滤效率)%(与N系列口罩测试标准不同) | - | - | 98 | E | |
巴基斯坦
以下就巴基斯坦医疗器械监管部门、相关法律法规,及进口产品用于销售的情况下,所需满足的进口资质、清关材料及相关产品标准等内容进行剖析。
监管机构
巴基斯坦药品监督管理局(Drug Regulatory Authority of Pakistan, DRAP)医疗器械和医疗化妆品司是巴基斯坦卫生服务、条例和协调部(Ministry of Health Services, Regulations and Coordination)下属的监管医疗器械进口、生产和销售的重要部门。
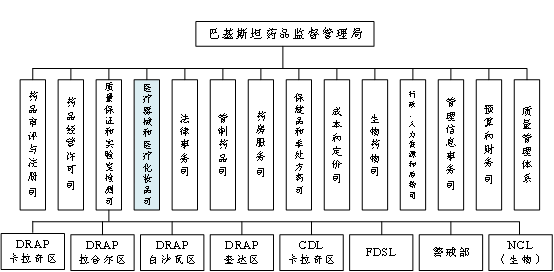
图3:巴基斯坦药品监督管理局组织机构图
法律法规
2018年1月,巴基斯坦药品监督管理局发布《医疗器械法规(2017)》(Medical Devices Rules, 2017),对医疗器械的分类、生产、进出口、销售、标签、广告等要求进行了规范。依据法规,医疗器械和医疗化妆品司成立了医疗器械委员会(Medical Device Board),负责管理医疗器械的登记或注册、颁发机构许可证、签发医疗器械或附件、零件进出口许可证等工作。
《医疗器械法规(2017)》规定,根据医疗器械的风险水平将医疗器械划分为A、B、C、D四大类别,依次代表低风险、中低度风险、中高度风险和高风险水平。其中,用于临时使用的检查用手套(examination gloves)、一次性口罩、药棉等属于A类;外科手术手套(surgical gloves)、电子体温计(Electronic thermometer)等属于B类。制造商需根据医疗器械的结构特征、使用形式、使用状况,确定产品所属类别。
清关流程与材料
1、进口医疗设备的单位应具备的资质
(1)医疗器械生产许可证或医疗器械经营许可证;
(2)营业执照(经营范围里有销售医疗器械许可);
(3)进出口权。
2、医疗器械进口报关需要准备的资料
(1)巴基斯坦药品监督管理局颁发给报关公司的进口医疗器械或附件、零件的登记或注册证;
(2)设备的检测报告复印件;
(3)经医疗器械委员会或巴基斯坦药品监督管理局官员背书的书面承诺;
(4)进口合同、3份发票复印件和自药品监督管理局获得的清关证明(即表11 进口医疗器械或附件、零件、原材料的到货通知单);
(5)其他需要补充的资料。
相关标准
目前,巴基斯坦国家标准中,防护用品相关标准(见表3)数量较少,且多采用国际标准,并均属于推荐性国家标准。2018年发布的《医疗器械法规(2017)》中明确规定,医疗器械的检测标准应当符合国际标准。
表3:巴基斯坦防护用品标准
类别 | 标准号 | 中文题名 | 英文题名 |
防护服 | PS:ISO 13688-2001 | 防护服的一般要求 | Protective clothing -- General requirements |
PS:ISO 6529-2014 | 防护服-化学试剂防护-防护服面料液体和气体的渗透性测定 | Textiles – Protective Clothing – Protection against chemicals – determination of resistance of protective clothing materials to penetration by liquid and gases. | |
PS:ISO 6530-2014 | 防护服-对液态化学制品的防护材料抗液体渗透性的试验方法 | Textiles – Protective Clothing – Protection against liquid chemicals – determination of resistance of materials to penetration by liquids. | |
PS:ISO 22612-2014 | 防止传染介质的防护服-抗干微生物穿透性的试验方法 | Clothing for protection against infectious agents -- Test method for resistance to dry microbial penetration | |
护目镜 | PS:ISO 4007-77 | 个人用护目镜-术语 | Eye-protectors – Personal – Vocabulary. |
PS:ISO 4854/1981 | 个人用护目镜光学性能试验方法 | Optical test methods – Personal eye-protectors. | |
PS:ISO 4855/1981 | 个人用护目镜非光学性能试验方法 | Non-optical test methods- Personal eye protectors. | |
手 套 | PS 698/1968 | 外科用橡胶手套 | Surgical Rubber Gloves |
PS 802/1986 | 验尸用橡胶手套 | Post mortem Rubber gloves (1st Rivision) | |
PS 4397/1999 | 一次性使用无菌手术手套 | Single use Sterile Surgical Gloves | |
PS 4744/2001 | 一次性使用橡胶检查手套 | Single use Rubber examination Gloves | |
其他 用品 | PS: IEC 60601-1/2014 | 医用电气设备第1部分:基本安全和基本性能的通用要求 | Medical electrical equipment. Part 1. General requirements for basic safety and essential performance |
PS:ISO 14708-1:2000 | 外科植入物-有源可植入医疗装置-第1部分:安全、标记及制造商提供信息的一般要求 | Implants for surgery -- Active implantable medical devices -- Part 1: General requirements for safety, marking and for information to be provided by the manufacturer | |
PS:ISO 22610-2014 | 医护人员和器械用手术单、手术衣和洁净服-测定防潮湿细菌渗透的试验方法 | Surgical drapes, gowns and clean air suits, used as medical devices, for patients, clinical staff and equipment -- Test method to determine the resistance to wet bacterial penetration |
孟加拉国
监管机构
孟加拉国药品监督管理总局(Directorate General of Drug Administration,DGDA)隶属于孟加拉国卫生和家庭福利部,其职责是对医疗器械的生产、进口、销售等进行监督管理,并确保药品质量、疗效和安全,以最大程度满足国民所需。
在孟加拉国,医疗器械分为A类(低风险)、B类(中低风险)、C(中高风险)、D类(高风险),根据2015年颁布的《医疗器械注册指南》,防护用品分类见表4。
表4:孟加拉国防护用品监管类别
防护用品 | 监管类别 |
医用外科口罩 | A类 |
隔离衣、医用帽 | A类 |
医用外科手套 | B类 |
隔离眼罩 | A类 |
医疗器械上市前批准程序由生产商和公告机构负责,A类产品无需从DGDA获得制造许可证,但生产商需要进行符合性声明并向DGDA报备;B、C、D类产品的设计与生产需要公告机构出具认证,生产商和进口商必须申请许可以及获得产品性能、安全方面的支持文件。
法律法规
DGDA按照以下法规条例的规定来行使职权:
《药品法案,1940》;
《药品条例,1945》及其修正案;
《药品条例,1946》及其修正案;
《药品(管制)条例,1982》及其修正案;
《药物(管制)条例修正案,2006》。
清关流程与材料
根据2015年颁布的《医疗器械注册指南》,对于首次进口或生产的医疗器械,申请人必须在进口或生产前向DGDA申请注册。费用在申请时一并提交,注册将由DGDA签发。
注册申请须由医疗器械生产商的授权人、当地授权代理商或者外国供应商按照规定的格式向DGDA提出,对于在同一场所生产的同类医疗器械,可以用一张申请表,但不得多于5种产品,如多于5种产品,应另外进行单独申请。
对于B、C、D类产品,公告机构须首先进行质量管理体系(QMS)认证,已在孟加拉境外获得由国家监管机构/公告机构颁发的QMS认证、产品认证或者销售授权的生产商,需要将相关资料提交DGDA;对于D类产品,还需对设备的设计进行认证,再由生产商或进口商连同DGDA规定的以下文件一并提交注册:
1)本地授权代理商的姓名、地址、电话、邮箱;
2)生产商授权本地代理商代为提交申请的授权书原件。如申请是由孟加拉国当地生产商的办事处提出,则不需要;
3)负责产品上市的公司或人员的名称、地址、电话号码和邮箱,如与生产商不同,则需生产商法人的证明文件;
4)如需在孟加拉国开展部分生产活动,则需提供当地生的商的详细信息;
5)产品的品牌、设备、通用名称;
6)按照全球医疗器械协调工作组(GHTF)分类的设备类别;
7)设备详细信息和说明;
8)设备尺寸;
9)设备的主要用途;
10)设备主文件,包括结构材料和定量分析细节(仅在CE或者FDA认证不适用时提交);
11)制造过程的简短描述,可提供多设施制造的详细信息,包括制造工艺简要说明和流程图;
12)标签和包装细节;
13)使用设备时所需附件的详细信息(如适用);
14)使用的标准(以ISO、ASTM、IEC、AAMI的标准优先);
15)设备使用说明书。
另外,注册申请时还需要提供生产国和在其他发达国家的监管情况,B类产品需提供原产国出具的自由销售证明(FSC);C类和D类产品,需提供欧盟、美国、加拿大、澳大利亚和日本中任一区域或国家出具的FSC,以及原产国FSC,还必须提交的是合格评定证书或同等资历证书,以及过去2年内因任何原因发生的任何退市或进行召回的详细信息(如有)。
孟加拉国进口业务一般情况下必须以不可撤销的信用证支付。除非孟加拉国商务部特许,进口贸易均不得采用CIF方式,以保护孟加拉国保险业及运输业。孟加拉自2002年2月15日开始实施进口商品“PSI—船前检验制度”(Pre-Shipment-Inspection),对输往孟加拉国的商品在装船前实施货值评估及散装状态检验。PSI属于强制性进口管理规范,一般由孟加拉国政府签约的国际商检机构进行装船前检查。商检机构完成检验后,在货物发票、箱单上背书,进口商凭此才能输报关等进口手续。对于进口到孟加拉国的产品,货运代理商需向孟加拉国海关当局提交电子版舱单数据(包括通过船舶进口的产品的说明)。如通过卡车运输经陆路关口,应由卡车公司或司机向海关提交进口货物清单(IGM)。IGM提交以后,指定的C&F代理或进口商需完成货物申报(俗称为入境单或提单),并通过海关数据自动化系统提交货物申报。申报或提单必须以一种特定的格式输出,称为单一行政文件(SAD)。其他需要报送的资料还包括信用证、发票、提单/运输收据、装箱单、“原产国”证书、保险单、增值税/银行标识代码证书。
对于医疗器械类产品,进口关税税率在0-25%区间。上述材料提交后经由海关评估,进口商或其C&F代理支付关税和税款,完税后,海关签发通关放行单,完成口岸手续后,货物清关。
B、C、D类医疗器械在孟加拉国上市后,生产商或进口商应遵守相关监管要求。严重不良事件应在知晓事件发生后10个工作日内向DGDA的指定机构提交分析报告,并应采取适当的纠正和预防措施,以防止或降低不良事件再次发生的可能性。
相关标准
截至目前,孟加拉国暂未制定医用防护用品相关国家标准。
尼泊尔
监管机构
1979年尼泊尔政府成立了药品管理局,现隶属于卫生与人口部,分别在Biratnagar,Birgunj和Nepalgunj成立了区域办事处。尼泊尔采购防疫物资是通过人口与卫生部组织国际招投标。现在尼泊尔药品管理局(DDA)注册的药品有11812种,药房21651家,进口商140家,生产商384家。
2020年3月23日尼泊尔药品管理局(DDA)根据2017年颁布的《不同药物特别许可须知》的相关规定对洗手液注册程序启动特别许可。
1978年,尼泊尔颁布了《药品法》,禁止滥用或滥用药物和相关药品以及与药物功效和使用有关的虚假或误导性信息,并规范和控制生产,销售,分销,出口,进口,储存和使用对公众不安全,有效和标准质量的药物。尼泊尔药品管理局(DDA)职责是规范与药物相关的,例如乱用和滥用药物及其原料,制止虚假和误导性广告。尼泊尔药品管理局(DDA)通过控制药品的生产,营销,分销,销售,出口,进口,储存和使用,向公众提供安全,有效和优质的药品,主要活动如下。
表5:尼泊尔药品管理局(DDA)职责范围
序号 | 活动 | 范围/职责 |
1 | 确保药物的安全性,质量和功效 | 药品和健康产品的评估,注册和许可。 颁发临床试验许可证,药房许可证以及推荐设立药品生产加工单元。 药品和健康科技产品进入市场后的监督。 检查和监视从事药品和健康科技产品的生产,销售,储存和分配。 注册前后药物分析和HTP测试。 |
2 | 促进合理使用药物和健康科技产品 | 制定和修订国家基本药物清单。 制定和修订抗生素处方管理办法。 向消费者传播有关合理使用药物的信息。 |
3 | 确保药品和健康科技产品享用权 | 制定药物政策确保获取药物享用权。 价格监管和价格透明。 颁发稀缺药品(包括孤儿药即“罕见药”)许可证。 |
4 | 强制执行毒品法规定 | 开展医学法律宣传活动。 制定有关药物注册,药房运营(包括医院药房)的标准和指南。 培训药品检查人员。 |
5 | 机构发展 (包括人力资源) | 审查组织状况并制定机构发展计划。 编写各级监管和分析人员的培训材料。 开展能力建设(例如人力资源建立/建设,改革和培训)。 为从事药品和健康科技产品制造,销售,储存和分配的公共和私营部门专业人员制定能力提升计划。 开发透明,响应迅速的服务交互系统(包括在线注册,网页和交流)。 |
尼泊尔药品管理局(DDA)的分支机构如下:
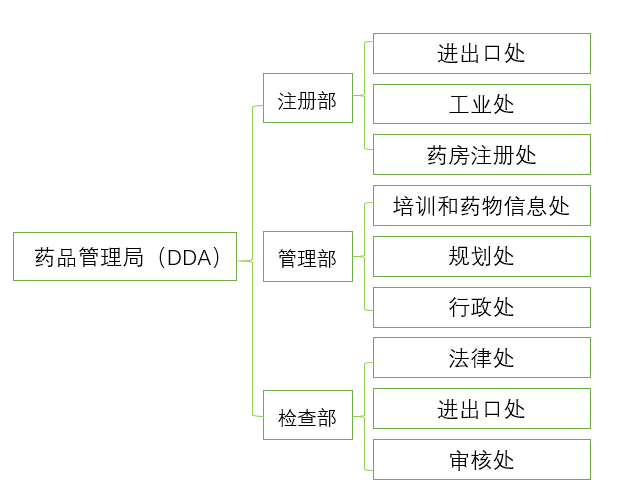
图4:药品管理局组织结构图
尼泊尔药品管理局(DDA)下设尼泊尔国家药品实验室,以前称为皇家药物研究实验室(RDRL),成立于公元1964年。它是尼泊尔政府进行药物测试和分析的主要机构,具有化学分析,微生物学,药理学和仪器分析等部分。NML的主要负责:测试和分析授权的药品质量;检查并评估国内药物测试实验室的标准;制定参考标准,并提供给制药行业和实验室;实验室规范培训;制药行业实验室审计。
尼泊尔卫生部下设物流管理科(LMIS),负责尼泊尔,省和地方政府的医疗机构计划,量化,采购,存储和分发。此外,还涉及生物医学设备,仪器和运输工具的维修和保养。LMIS每季度(每月三个)收集并分析来自全国所有卫生机构的报告,预测公共卫生计划商品的年度需求,包括计划生育,孕产妇,新生儿和儿童健康,艾滋病毒和艾滋病商品,疫苗和基本药物;帮助确保各级药物,疫苗,避孕药具和基本医疗用品的需求和供应; 季度监测国家管道和主要卫生商品的库存水平。
尼泊尔标准理事会,其缩写为N.C.S,尼泊尔标准(合格评定)法案规定了理事会的职能,职责和权力,以及尼泊尔标准局建立职能,职责和权力,名称以及简写;使用合格评定标志必须遵守该法案。所有特定数量商品的标准认证都需向办事处提出申请,进行调查和确认商品质量。由办事处指定工作人员对商品进行检查。
尼泊尔人口与卫生部公布的国际招投标个人防护用品中,第22项GUN BOOT获得需求,规定该产品需要获得ISI认证。尼泊尔国际招投标《Procurement of Different items For Covid-19 Control and Prevention Procision Cencering Procurement to be Made in Special Circumstances》中标准和安全要求6.1规定需获得CE(93/42EEC Directives)或者美国FDA产品认证证书。尼泊尔标准计量局规定制造商自身或其指定代理机构出具的质量保证/测试证书。

法律法规
医疗设备指令中涉及医疗设备所属的类别I类(低风险,low risk),IIa类(中等风险,medium risk),IIb类(中等风险,medium risk),III类(高风险,high risk)。属于I类(低风险)的制造商应自行进行合格性评估并记录评估结果,但需有CE认证标志。IIa类(中等风险,medium risk),IIb类(中等风险,medium risk)和III类(高风险,high risk)设备中非定制或计划用于临床研究,除加贴CE标志外,制造商应进行合格评定并进行评估,并附上医用设备技术法规(93/42EEC)中规定的以上3类所需的其他文件。
通关相关法律法规主要包括《海关法(2007)》、《海关规则(2007)》等。《海关法(2007)》是尼泊尔管理进出口关税及规范关税估价标准的主要法律依据。该法共14章,主要内容有:海关区域及办公场所、关税、关税便利化、海关估价、申报、检验、通关、验货、事后稽查、搜查和拘留、货物查封、没收和拍卖、清关代理、处罚、复议和上诉等。《海关规则(2007)》主要内容有:关税免除优惠、保税仓库、银行担保、特别经济区、申报检查、货物没收及审计程序、海关估价复议、废弃物销毁、清关代理、事后稽核等。此外,尼泊尔标准与计量局执行产品认证计划以保证尼泊尔工业产品质量,该计划是一个自愿性计划。通过此认证计划的产品可以获得尼泊尔标准认证标志。目前,126个行业中的55类产品已经被授予尼泊尔标准认证标志。除药品外,尼泊尔品牌产品不需要产品认证。对于进口药品,进口商必须事先从药物管理司获得批准。药品的化学成分已在药物管理司获批的情况下,被认可的制造商生产的专利药品不需要认证。食品产品、易造成海关分类混淆的化工材料,海关部门必须对其质量标准进行检验的项目、药品原料等海关货物需在实验室进行检测。
尼泊尔进出口商品检验由尼泊尔标准计量局按照《尼泊尔标准(证明标志)法(1980)》规定完成。
清关流程与材料
在尼泊尔海关通关有两种形式,一是进出口商直接向海关申报办理通关手续;二是进出口商委托报关代理行办理通关手续。尼海关对报关代理行每年进行一次年审换证。
(1)进口通关程序
A、货物运抵港口或边境口岸海关,进口商或报关代理向海关报关时提交下列文件:
进口报关单、提货单、商业发票、装箱单、运输单据、原产地证、保险单、银行出具的信用证复印件或换汇单、公司营业执照复印件、公司交纳所得税证明及交纳增值税证明、商检证,以及要求报关行代理的委托书。
B、海关官员审核上述文件无误后便由海关指定检查人员和海关人员对货物进行查验。查验无误后进口商按规定交纳关税和增值税(10%)及消费税(1.5%),海关会计部门签发收据,进口货物便清关放行。
C、上述文件中的提货单、商业发票、装箱单、运输单据、保险单、银行出具的信用证复印件或换汇单等对海关确定进口商品的价值十分重要。尼泊尔海关备有专用的海关商品价格清单,原则上该清单每6个月更换一次。如查验人员认为进口货物价格与清单上的价格有较大差别,将参照清单上的商品价格进行征税。对清单未列商品将参照类似商品征税,或提交评估委员会进行评估。另外,尼泊尔海关除征收关税、增值税、消费税外,还将根据所进口商品与尼泊尔国内生产的同种商品纳税税率征收一种平衡税。
相关标准
尼泊尔国内执行尼泊尔标准(NS),其国家标准可以分为两类:一是尼泊尔本国自行制定的标准;二是采用外国/国际标准。通过官网收集资料暂未见尼泊尔口罩、医用防护服、护目镜、消毒液等防护用品国家标准。疫情期间尼方已向保持友好关系的中国提出了防护用品标准需求。经过协商,我国已向尼泊尔提供了24项防疫物资相关标准题录,包括7项口罩标准题录,2项防护服标准题录,1项护目镜标准题录,6项手套标准题录,8项消毒剂标准题录。
不丹
监管机构
成立于2004年6月的不丹药品监督管理局(Drug Regulatory Authority, DRA)是不丹卫生部下属的监管药品和医疗器械进口和销售的重要部门。在制定政策法令方面,不丹药监局直接向不丹药品委员会(Bhutan Medicines Board)报告。药品技术顾问委员会(Drug Technical Advisory Committee, DTAC)和血液技术顾问委员会(Blood Technical Advisory Committee ,BTAC)为药监局提供技术支持。药监局下辖三个技术部,注册部,检查部,市场监管部,这三个部门的工作由管理部和财务部支持。
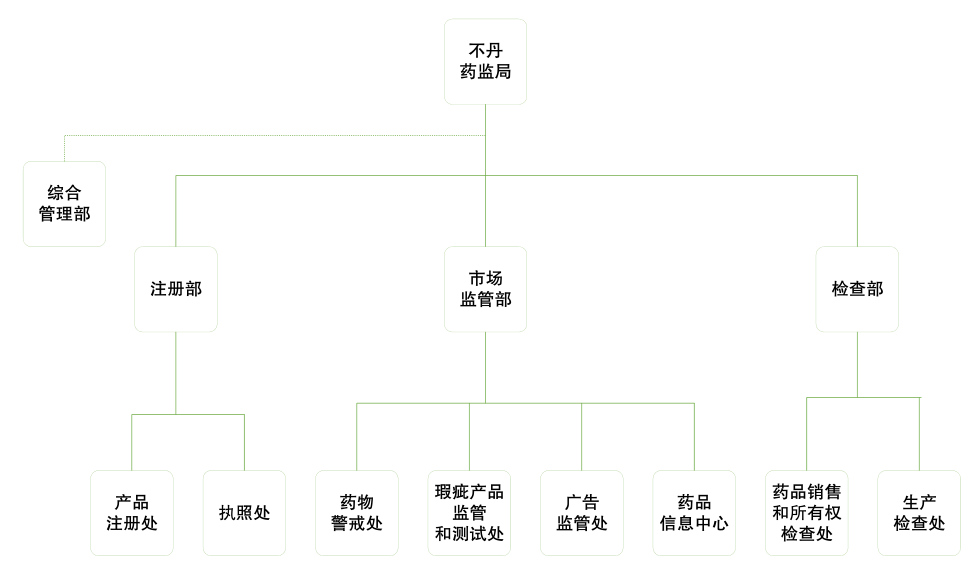 图5:不丹药品监督管理局组织架构
图5:不丹药品监督管理局组织架构
不丹药监局的主要职能:
-授权制造,进口,出口,销售,分销和储存包括血液和血液制品在内的药品。
-负责国内生产或进口到该国的药品注册。
-监管从事药品进口,储存,制造和销售的人员的能力和技能。
-对生产,销售,分销和储存包括血液和血液制品在内的医药产品的场所进行检查/监管。
-以不丹国家配方和药物安全信息的形式提供最新信息。
-监测由药品引起的不良反应的趋势和情况。
-向公众介绍药品的使用和有害作用。
-促进政策,以改善对具有成本效益的优质药品的获取。
-对与药品有关的问题进行研究。
-以经济有效的方式向公众提供监管服务。
法律法规
现阶段不丹还没有明确的医疗器械法规来监管产品的安全、质量和性能。根据不丹药品委员会(Bhutan Medicines Board)的指示,不丹药监局正在制定医疗器械法规(Medical Device Regulation)。
根据不丹药品监管局(Drug Regulatory Authority,DRA) 2018年6月发布的Medical Device Control Strategy 2018-2023, 2019年6月,DRA打算制定并出台一份医疗器械法规(Medical Device Regulation,MDR)。实际情况是截至2020年3月份至今还未出台。不过在MDR法规发布之前,不丹药监局将采用全球医疗器械协调工作组(GHTF)制定的标准,以及后来的国际医疗器械监管者论坛(IMDRF)制定的医疗器械标准。
根据正在制定的医疗器械法令(MDR),不丹药监局打算将医疗器械按风险由低到高分为A类、B类、C类、D类共四级。例如测温仪属于A类,心脏起搏器属于D类,不过未说明防疫产品属于哪一类。A类产品不需要注册,主要通过进口授权和上市后监管。B类,C类,D类需要注册,也通过进口授权和上市后监管。
现有药品相关的监管法令如下:
- 药品法案,2003
- 不丹药品法规法令2019
- 国家药品政策2007
- 不丹血液及血液制品法令2016
清关流程与材料
在不丹,注册证书和进口许可证是进口药品和医疗器械的强制性要求。
注册证书:要注册药品,首先当地经销商(称为市场授权持有人Market Authorization Holder)或制造商应向DRA申请销售和分销技术授权(Technical Authorization for Sale and Distribution),经济事务部下属的贸易司将根据该授权签发商业许可证(Trade License)。获得销售和分销的商业许可证和技术授权后,当地经销商可以将与药品有关的文件提交DRA进行注册。如果不丹以外的制造商希望直接注册药品,也可以这样做。如果相关资料准备充分,大约1-3个月时间可以完成,注册证书有3年有效期。
如果药品在如下国家药品监管机构完成了注册,进入不丹可以免除注册:澳大利亚的TGA, 加拿大的HC, 美国FDA, 欧盟EMA, 英国MHRA, 日本DRA,新加坡HSA,马来西亚BPFK,泰国FDA.或是由WHO,UN, OIE或其它联合国机构预认证过的药品及疫苗。
进口许可证:药品与医疗器械在不丹属于进口管制产品,进口的时候需要获得药监局颁发的进口授权(Import Authorization)。进口授权只能颁发给市场授权持有人(market authorization holder),不丹当地注册的药店,政府采购部门,国际组织或是由药监局DRA授权的个人。
不丹主管进口的部门为经济贸易部(Ministry of Economic Affairs)下属的贸易司(Department of Trade)。在不丹,获得注册证书和进口许可证是进口药品和医疗器械的强制性要求。
在进口产品到达不丹之前,进口商或是其授权代理人需向位于印度加尔各答的不丹海关联络中转办公室( the Bhutanese Revenue and Customs Liaison and Transit Office)提交如下资料:
1.进口许可证副本;
2. 海运提单/空运提单/装船清单,需由进口商和相关银行盖章;
3. 发票;
4. 装箱单;
5. 原产地证;
6. 海运保险/空运保险/陆运保险。
货物到达不丹7个工作日内,进口商或是其授权代理商应完成清关手续。清关时,进口商或是其授权代理商需提交进口声明表,主要包含内容如下:
1. 进口声明表应包括进口产品的详细信息,进口商需保证其真实性,并附上货运单据及银行盖章的发票。
2. 如果有必要,海关官员应该对进口产品进行实物检查。
3. 如果海关官员检查产品没有问题,在进口商交付了关税、销售税及其它费用之后,就应该发出放行指令。
相关标准
不丹卫生部下辖的医疗用品和卫生基础设施部(Department of Medical Supplies and Health Infrastructure,DoMSHI)下设的生物医学工程处(Bio-Medical Engineering Division,BMED)采用了50多项医用电气设备IEC和ISO标准做为国家标准。与防疫相关的标准涉及医用体温计及脉搏血氧仪标准,这两个标准等同采用了ISO标准,具体情况如下表6。不过不丹标准局(Bhutan Standards Bureau)也在与相关利益方合作制定本国的医疗器械标准。
不丹药监局可接受的标准有:国际标准(WHO,ISO,IEC,GHTF,IMDRF等)、印度BIS标准、欧美主要发达国家(FDA等)。目前,不丹政府不接受中国标准(包括国家标准、行业标准、企业标准等)。
表6:不丹防疫产品标准(等同采用ISO标准)
标准号 | 标准中文名 | 标准英文名 |
BTS 109:2018 ISO 80601-2-56:2017 | 医用电气设备-第2-56部分:医用体温计的基本安全与基本性能的特殊要求 | Medical electrical equipment - Part 2-56: Particular requirements for basic safety and essential performance of clinical thermometers for body temperature measurement |
BTS 110:2018 ISO 80601-2-61:2017 | 医用电气设备-第2-61部分:脉搏血氧仪的基本安全与基本性能的特殊要求 | Medical electrical equipment -- Part 2-61: Particular requirements for basic safety and essential performance of pulse oximeter equipment |
斯里兰卡
监管机构
根据2015年第05号《国家药品监督管理局法案》,口罩、手套、防护鞋服等医疗器械产品的注册,许可,制造,进口和其他监管活动,均由隶属于斯里兰卡卫生部的国家药品监督管理局(National Medicines Regulatory Authority, NMRA)负责。NMRA旨在确保国内药品符合相应安全,质量和功效的适用标准,在保护和改善国内公共健康方面发挥着领导作用。国家药品监督管理局监管范围可分为4类:药品,医疗器械,边界产品,临床试验和化妆品。其下设的医疗器械评估委员会(Medical Device Evaluation Committee, MDEC)和国家药品质量保障实验室(National Medicines Quality Assurance Laboratory, NMQAL)分别负责口罩、手套、防护鞋服等医疗器械产品的准入登记和产品检测。
医疗器械评估委员会(Medical Device Evaluation Committee, MDEC)根据相应医疗器械的质量,安全,有效,需求以及成本,对注册的产品进行技术评估。MDEC会定期组织医学和制药领域的专家,讨论和评审药品营销许可申请,并制定与药品营销许可有关的政策。
国家药品质量保障实验室(National Medicines Quality Assurance Laboratory, NMQAL)负责提供药品,医疗设备,边界产品和化妆品的质量检测技术支持。NMQAL在进行质量、安全评估时遵循标准药典中的测试程序以及其他公认的(经过验证的)测试程序。NMQAL由生物部、化学部、微生物部和技术部组成。
法律法规
2015年斯里兰卡颁布第5号法案,《国家药品监管局法》(NATIONAL MEDICINES REGULATORY AUTHORITY ACT)废除了1980年《化妆品、器械和药品(CDD)法》。《国家药品监管局法》是斯里兰卡现行国内化妆品、药品以及医疗器械的立法框架,并为相关监管活动提供了法律依据。斯里兰卡国家药品监督管理局负责贯彻实施该法案。
清关流程与材料
口罩、手套、防护服等医疗器械产品制造商均需要通过斯里兰卡的销售授权持有人(当地代理商)在NMRA提交产品注册申请。该注册人应负责一般医疗产品在斯里兰卡注册,许可,进口,销售和分销等一系列活动,同时还需要处理不合格产品以及与特定医疗有关的注册和许可申请。产品在国家药品监督管理局注册登记申请通过后,方可获得进口许可,进入斯里兰卡市场。
1、准入流程
大致来讲,针对尚未在NMRA注册的口罩、手套、防护鞋服等医疗器械,斯里兰卡准入流程主要为:授权进口销售商、提交样品进口许可申请、提交产品注册申请、获取进口证书、销售产品。
 图6:口罩、手套、防护服等医疗器械产品准入流程
图6:口罩、手套、防护服等医疗器械产品准入流程
表7:斯里兰卡药品及医疗器械注册和许可要求
监管机构 | 国家药品监督管理局(NMRA) |
网址 | http://nmra.gov.lk/ |
注册时间 | 3个月 |
注册有效期 | 首次注册将签发为期一年(或两年)的临时注册,并在证书中指定;产品的完全注册有效期为五年,并在证书中指定。 |
进口许可证有效期 | 1年 |
本地代理/子公司的要求 | 不需要子公司。经授权的本地进口商可以进行注册。 |
工厂检查 | 无强制性要求 |
2、样品进口许可证申请流程及所需材料
(1)口罩、手套、防护服等医疗器械产品出口到斯里兰卡首先需要获得斯里兰卡卫生部(Ministry of Health , MOH)颁发的样品进口许可证,具体步骤如下所示。
 图7:医疗器械注册涉及的步骤
图7:医疗器械注册涉及的步骤
(2)申请样品许可证,申请人应提交:
a)《化妆品、器械和药品条例附表四》;
b)申请人的商业登记证副本,应注明董事会、秘书委员会的详细情况;
c)制造商指定的斯里兰卡当地代理商授权书;
d)原产国卫生局提供的特定产品的免费销售证明复印件;
e)计划销售的完整包装样品(两个)(包括批号,制造日期,截止日期,制造商和进口商的详细信息)
3、NMRA注册步骤及所需材料
(1)在获得样品进口许可证后,口罩、手套、防护服等医疗器械产品当地代理商就可在NMRA进行产品注册,具体注册步骤如下所示。
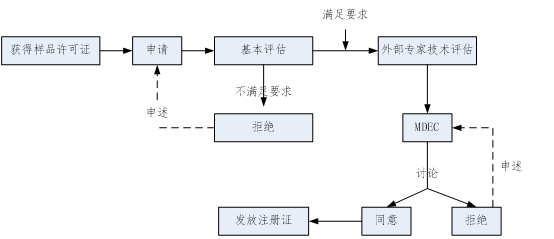 图8:口罩、手套、防护服等医疗器械在NMRA的注册步骤
图8:口罩、手套、防护服等医疗器械在NMRA的注册步骤
(2)申请注册产品,申请人应提交的基本文件如下:
a)索引;
b)确认函;
c)进口许可证样本副本;
d)《化妆品、器械和药品条例附表I-附表A》:申请人的名称和地址、设备的制造商和进口名称。品牌名称(如有)、官方或经批准的名称。生产国卫生当局出具的证明,确认设备在该国使用和使用期限,如果没有,需要说明不在制造国销售的原因(免费销售证书)。由FDA/医疗器械控制机构/斯里兰卡大使馆或外交部认证或D/CDD认可的国外机构颁发的免费销售证书原件或副本。已在其他国家进行注册的国家名单,如国外注册证书。设备的全包装样品。
e)申请人应能提交至少两份商业批次的样品,以便发送给外部评估人员。仪器仪表也应提交两份样品。
需要注意的是:填写的《化妆品、器械和药品条例附表I-附表A》需要与表格中的要求文件一起提交。文件应为英文,字体清晰易读,A4打印,并以硬文件封面(Box文件)提交,所有页面均应从上至下编号,并带有索引。如果有关主管当局以任何其他语言签发了原始证书或许可证,则应提交经认证的英语翻译。
注册申请必须完整且按照规范处理。
应针对每个要注册的设备分别提出申请。外国制造商的产品应通过营销授权持有人提交。
除上述基本文件/细节外,如有必要,还应提交下列技术文件:
a)如纱布、绷带、手术手套、医用气瓶等需要斯里兰卡标准协会(SLSI)的检测报告。
b)缝合线,镊子,剪刀等医疗器械需要材料试验报告。
c)提交ISO 13485认证材料,以便NMRA对制造商的设计、开发、制造以及上市后安全和性能进行监控。
d)CE认证,如果有在欧盟地区销售。
相关标准
标准号 | 标准名称-中译文 | 标准名称-英文 |
SLS 1626 | 一次性无菌橡胶手术手套 | Single-use sterile rubber surgical gloves |
SLS 1623 | 通用一次性橡胶手套 | Single-use rubber gloves for general applications |
SLS 688 | 消毒剂 | Disinfectants |
SLS 1363 | 个人防护鞋测试方法 | Methods of test for personal protective footwear |
SLS 1626: Single-use sterile rubber surgical gloves(一次性无菌橡胶手术手套)规定了外科手术中使用的无菌橡胶包装手套要求,以保护患者和用户免受交叉污染。该标准适用于一次性手套,不适用于检查或操作手套。一次性无菌橡胶手术手套用光滑的表面覆盖,并在部分或整个手套上设有纹理表面。该标准为橡胶外科手套性能和安全性提供技术参考。手术手套和灭菌程序以及随后的搬运、包装和储存程序的安全和正确使用不在本标准范围内。SLS 1626等同采纳ISO 10282: 2014。
SLS 1623:Single-use rubber gloves for general applications(通用一次性橡胶手套)规定了由天然橡胶胶乳、合成橡胶胶乳或橡胶溶液制成的一次性橡胶手套的物理要求和取样和试验方法,这些手套适用于一般用途,但不适用于医疗用途。SLS 1623等同采纳ISO 25518: 2009。
SLS 688:Disinfectants(消毒剂)规定了消毒剂的取样和试验要求和方法。
SLS 1363:Methods of test for personal protective footwear(个人防护鞋测试方法)规定了作为个人防护装备设计的鞋类的试验方法。













